The Sharifi laboratory investigates how hormone physiology shapes therapeutic resistance through various mechanisms including non-canonical steroid biosynthesis, local hormone exposure, and inherited metabolic variation.
Research Projects
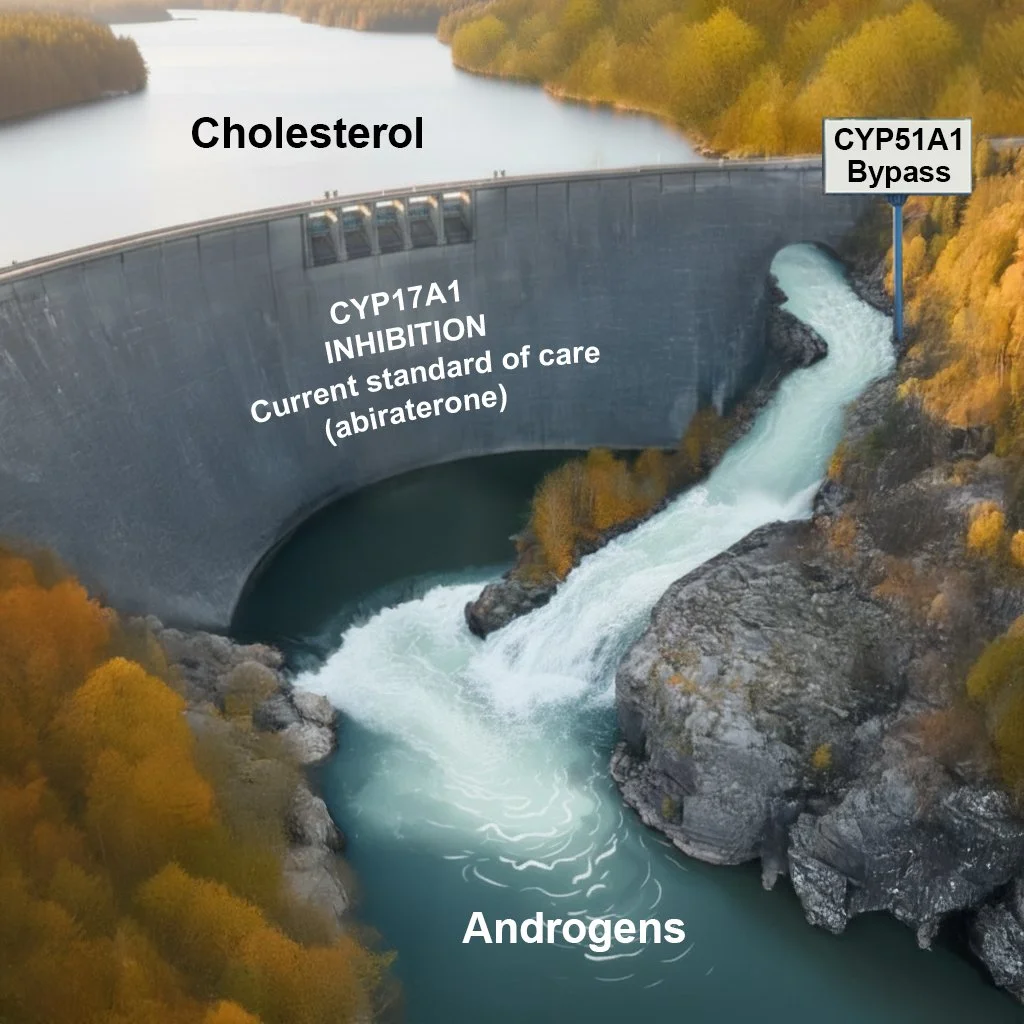
CYP51A1: A newly discovered CYP17A1-independent pathway for sex steroid biosynthesis
A major new focus of the laboratory is the discovery of a CYP17A1-independent pathway for androgen and estrogen biosynthesis mediated by CYP51A1, an enzyme previously believed to function exclusively in cholesterol biosynthesis. We demonstrated that CYP51A1 converts oxysterol intermediates derived from cholesterol into DHEA, testosterone, and dihydrotestosterone, bypassing the canonical requirement for CYP17A1 and sustaining androgen receptor signaling even during pharmacologic CYP17A1 inhibition (Zhu, et al. Nature Communications 2025). Genetic ablation of CYP51A1 suppresses androgen signaling and tumor growth in castrated prostate cancer xenograft models, establishing CYP51A1 as a previously unrecognized regulator of sex steroid physiology.
This work has broad implications for resistance to androgen-targeted therapies and extends to estrogen biosynthesis, positioning CYP51A1 as a potential driver of hormone-responsive cancers beyond prostate cancer, including breast cancer. Current work focuses on further elucidating the CYP51A1-dependent steroidogenic pathway, developing pharmacologic strategies to target this enzyme, and determining whether CYP51A1 activity or associated metabolic signatures can serve as predictive or pharmacodynamic biomarkers in hormone-responsive cancers.
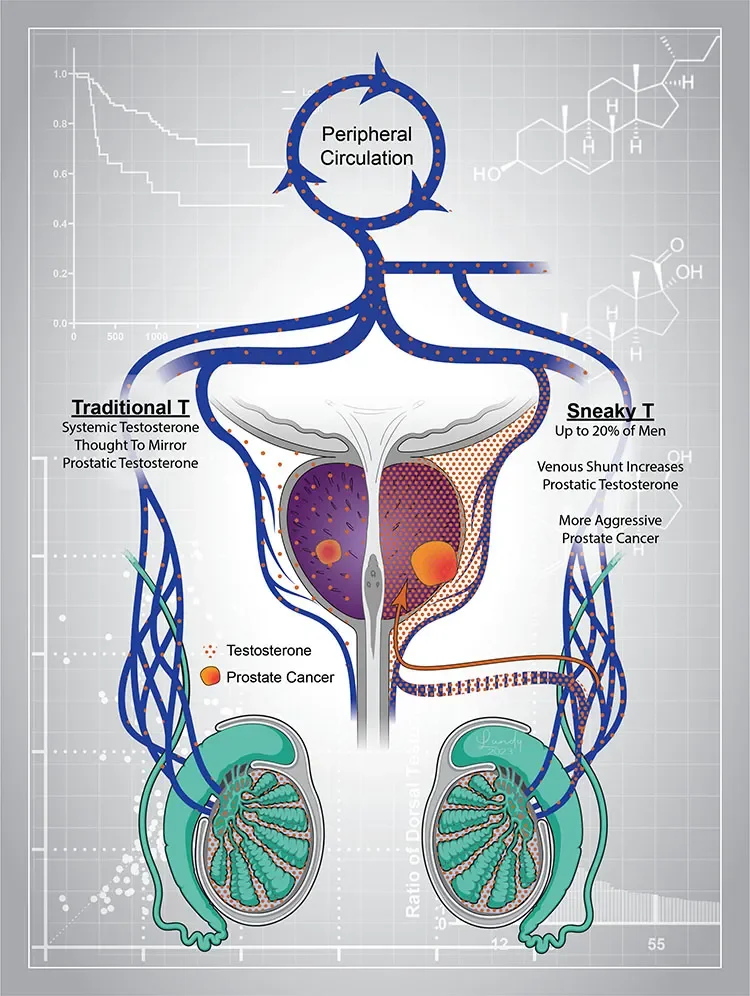
A short circuit from gonadal circulation to the prostate: “Sneaky T” physiology
We have identified a previously unrecognized form of androgen exposure in a subset of men with prostate cancer characterized by markedly elevated periprostatic venous testosterone concentrations that are not detectable in systemic circulation (Alyamani et al., J Clin Invest 2023). This localized testosterone likely originates from gonadal production and reaches the prostate via a short-circuit venous pathway. Men with this “sneaky testosterone” physiology experience worse oncologic outcomes following prostatectomy, suggesting clinically meaningful disconnect between serum testosterone and actual local prostate cancer androgen exposure. Current work focuses on defining the vascular and metabolic basis of this physiology, understanding its interaction with intraprostatic androgen biosynthesis pathways, and developing non-invasive diagnostic strategies to identify affected patients.
Genetic variation in androgen synthesis machinery and patient outcomes
We investigate how genetic alterations enable cancer cells to evade androgen deprivation therapy (ADT) by sustaining local hormone synthesis. Our laboratory identified the “adrenal-permissive” activity of the HSD3B1(1245C) variation, which encodes a hyperactive 3β-hydroxysteroid dehydrogenase-1 enzyme that accelerates conversion of adrenal steroid precursors into potent androgens (Chang et al., Cell 2013). This variation consistently predicts inferior outcomes and altered treatment responses across multiple clinical contexts, including ADT and is now known to be the most common inherited link to prostate cancer mortality (Hearn et al., Lancet Oncol 2016; JAMA Oncol 2018, 2020; Almassi et al., JAMA Oncol2018). We have also discovered that patients with the HSD3B1(1245C) variation metabolize abiraterone differently, producing androgen-like metabolites that paradoxically activate androgen receptor signaling and promote resistance (Li et al., Nature 2015, 2016; Alyamani et al., J Clin Invest 2018). Recently, we identified a previously unrecognized intestinal pathway linking inherited HSD3B1 variation to bile acid metabolism in prostate cancer patients receiving androgen deprivation therapy. This work expands the physiological significance of the HSD3B1 genotype, and the laboratory continues to investigate its roles in cancer and normal physiology (Fotouhi et al. J Clin Invest 2026).
Current work aims to validate these findings and identify alternative treatment strategies that overcome genotype-specific resistance mechanisms.
More broadly, we study steroid metabolism at the interface of inflammation and endocrine signaling. Systemic glucocorticoids suppress adrenal androgen production, an underappreciated effect with clinical consequences. We showed that HSD3B1 genotype predicts response to glucocorticoids in severe asthma, likely by determining how suppressed adrenal steroids are converted into bioactive androgens in peripheral tissues (Zein et al., PNAS 2020). This work highlights shared steroid metabolic pathways that shape therapeutic response across cancer and inflammatory diseases.

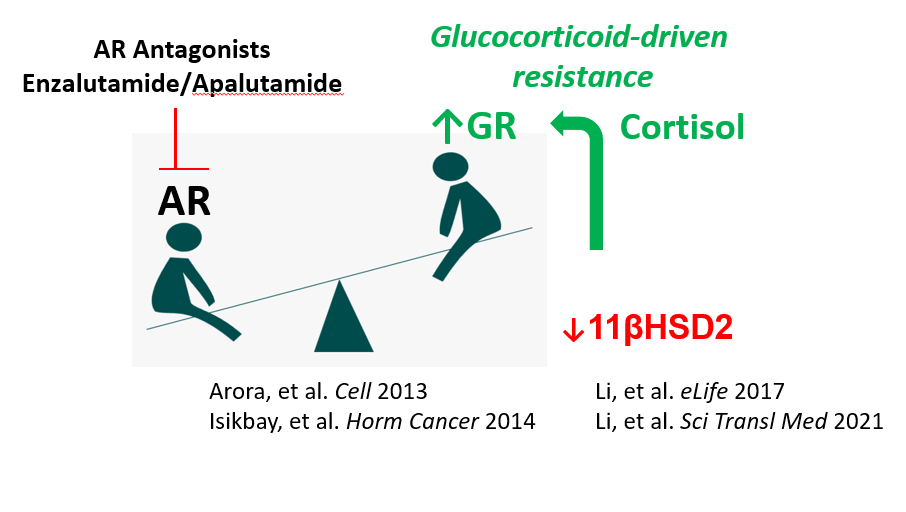
The androgen-glucocorticoid interface and aberrant glucocorticoid metabolism
Our laboratory has defined how prostate cancer develops aberrations in glucocorticoid metabolism that promote resistance to androgen receptor antagonists such as enzalutamide. Loss of cortisol-inactivating pathways results in elevated intratumoral glucocorticoids that substitute for androgens in driving resistance (Li et al., eLife 2017). We identified hexose-6-phosphate dehydrogenase inhibition as a strategy to reverse this resistance (Li et al., Sci Transl Med 2021). In parallel, we discovered that AR antagonists systemically disrupt glucocorticoid inactivation, increasing exposure to bioactive glucocorticoids and potentially contributing to treatment-related adverse effects (Alyamani et al., Ann Oncol 2020).